Химия и химические технологии / 5. Фундаментальные проблемы создания новых материалов и технологий
*Белевцова
Т.В., к.х.н. *Бондарев Н.В., к.х.н. **Зайцева И.С.
*Национальный
университет имени В.Н. Каразина, Харьков, Украина
**Харьковская
национальная академия городского хозяйства, Украина
Метод констант равновесий
и термодинамика химических превращений
Актуальной
задачей теории химических равновесий
является выявление термодинамических
закономерностей химических превращений в различных средах с целью
прогнозирования выхода продукта реакции и управления химическим процессом.
Цель
данной работы – рассмотреть основные источники ошибок определения энтальпии
(энтропии) химического равновесия эмпирическим дифференцированием и изложить
основы регрессионного анализа температурной зависимости констант равновесия для
оценки стандартных термодинамических характеристик химического превращения.
Зная температурную зависимость
константы равновесия можно рассчитать не только стандартную энергию Гиббса
процесса DrGo = –RT ln K, но и оценить другие термодинамические
параметры – энтальпию DrНo, энтропию DrSo и теплоемкость DrСо реакции, например, с помощью уравнения Вант-Гоффа ln K = A + B/T – для равновесий комплексообразования
или Харнеда-Робинсона ln K = A1/Т
+ A2 + A3T – для равновесий диссоциации. Это крайне важно
в тех случаях, когда прямым калориметрическим экспериментом определить DrНo невозможно (низкая растворимость реагентов, небольшие скорости процессов).
В то же время нахождение термодинамических характеристик реакции из температурной
зависимости константы равновесия (методом констант равновесия) сопряжено с
серьезными экспериментальными проблемами точности нахождения DrНo и DrSo, обусловленными общим температурным интервалом (Т2 – Т1) проведения эксперимента и ошибками определения
констант равновесия при разных температурах (табл. 1 и 2).
Таблица 1. Точность определения
DrHo с учетом ошибки dK в
константе равновесия и температурного интервала
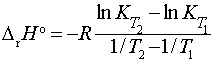 ,
, 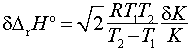 [1]
[1]
|
|
dlgK |
T2 – T1 |
|
кДж/моль |
|
0.05 |
±0.02 |
50 |
3002 |
±1.1 |
|
25 |
±2.2 |
|||
|
10 |
±5.3 |
|||
|
0.10 |
±0.05 |
50 |
3002 |
±2.1 |
|
25 |
±4.2 |
|||
|
10 |
±10.6 |
Таблица 2. Точность определения
DrSo с учетом ошибки dK в
константе равновесия и температурного интервала
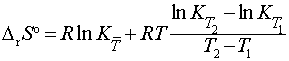 ,
, 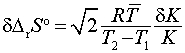 [1]
[1]
|
|
dlgK |
T2 – T1 |
|
Дж/(моль·К) |
кДж/моль |
|
0.05 |
±0.02 |
50 |
300 |
±3.5 |
±1.1 |
|
25 |
±7.0 |
±2.1 |
|||
|
10 |
±17.6 |
±5.2 |
|||
|
0.10 |
±0.05 |
50 |
300 |
±7.0 |
±2.1 |
|
25 |
±14.1 |
±4.2 |
|||
|
10 |
±35.2 |
±10.5 |
В работе предложен подход (на примере экспериментальных констант устойчивости
комплексов 18С6 с катионами калия K+ + L = LK+ в
водно-пропан-2-ольных растворителях [2]) к оценке
точности термодинамических характеристик комплексообразования (табл. 3),
основанный на регрессионном анализе температурной зависимости констант
равновесия; оценке значимости уравнения регрессии по критерию Фишера; оценке
значимости коэффициентов регрессии по критерию Стьюдента; табличном и
графическом анализе остатков, подтверждающем адекватность модели и нормальное
распределение остатков (показатель Кука, расстояния Махаланобиса).
Значимость уравнения регрессии lg K = A + B/T для аппроксимации температурной зависимости констант
равновесия подтверждена F–критерием дисперсионного анализа (Fнабл > Fкр)
Fнабл = ( r2
/k ) / [(1 – r2)/( n–k–1)]
при
проверке выполнимости нулевой гипотезы H0:
A =0, B = 0. Здесь n – число
экспериментальных точек, k – число
коэффициентов уравнения регрессии без свободного члена; r – коэффициент корреляции.
Проверка значимости коэффициентов
A и В проведена по критерию Стьюдента (![]() > tкр).
Гипотеза H0: A = 0,
> tкр).
Гипотеза H0: A = 0, ![]() ; гипотеза H0:
B = 0,
; гипотеза H0:
B = 0, ![]() , где sA
и sB – стандартные
отклонения коэффициентов уравнения регрессии. Доверительные интервалы: DrGо ±
2.303RTdlgKLM, DrНо ± 2.303tn-k-1sBR, DrSо ± 2.303tn-k-1sAR.
, где sA
и sB – стандартные
отклонения коэффициентов уравнения регрессии. Доверительные интервалы: DrGо ±
2.303RTdlgKLM, DrНо ± 2.303tn-k-1sBR, DrSо ± 2.303tn-k-1sAR.
Таблица
3. Параметры регрессионного анализа температурной зависимости констант
устойчивости коронатов (lgK = A
+B/T) и стандартные термодинамические характеристики комплексообразованияв в растворителях вода-2-PrOH
|
мас. доли спирта |
A (sA, tнабл) |
B (sB, tнабл) |
r |
s×102 |
lgKLK+, 298.15 |
DrG0 |
DrH0 |
TDrS0 |
n; Fнабл (Fкр; tкр), DT |
|
кДж/моль |
|||||||||
|
0 |
-2.37 (0.09; 26) |
1310 (27; 48) |
0.999 |
0.8 |
2.04 ±0.01 |
-11.7 ±0.06 |
-25 ±1.3 |
-13 ±1.3 |
7; 2265.8 (6.61; 2.57), 30 |
|
0.2 |
-2.18 (0.06; 37) |
1379 (17; 79) |
0.999 |
0.6 |
2.46 ±0.01 |
-14.0 ±0.06 |
-26 ±0.8 |
-12 ±0.8 |
8; 6257.0 (5.99; 2.45), 35 |
|
0.4 |
-0.83 (0.08; 10) |
1087 (25; 43) |
0.998 |
0.9 |
2.83 ±0.01 |
-16.2 ±0.06 |
-21 ±1.2 |
-5 ±1.2 |
8; 1859.2 (5.99; 2.45), 35 |
|
0.6 |
-2.93 (0.09; 32) |
1794 (28; 65) |
0.999 |
1.0 |
3.10 ±0.01 |
-17.7 ±0.06 |
-34 ±1.3 |
-16 ±1.3 |
8; 4261.5 (5.99; 2.45), 35 |
|
0.8 |
2.73 (0.04; 72) |
318 (11; 28) |
0.996 |
0.4 |
3.80 ±0.01 |
-21.7 ±0.06 |
-6 ±0.5 |
16 ±0.5 |
8; 771.9 (5.99; 2.45), 35 |
|
1.0 |
-0.19 (0.03; 7) |
1251 (8; 160) |
0.999 |
0.3 |
4.01 ±0.04 |
-22.9 ±0.06 |
-24 ±0.4 |
-1 ±0.4 |
8; 25230 (5.99; 2.45), 35 |
Литература:
1. Гаммет Л. Основы
физической органической химии. М.: Мир, 1972, 512 с.
2. Бондарев Н.В. ЖФХ, 1999, 73, 74–77